service@bersinbio.com
+86-20-32033089
Toggle Navigation

Time of publication:June,2024

Summary
Background: Lung adenocarcinoma (LUAD) patients have a dismal survival rate because of cancer metastasis and drug resistance. The study aims to identify the genes that concurrently modulate EMT, metastasis and EGFR-TKI resistance, and to investigate the underlying regulatory mechanisms.
Methods: Cox regression and Kaplan-Meier analyses were applied to identify prognostic oncogenes in LUAD. Gene set enrichment analysis (GSEA) was used to indicate the biological functions of the gene. Wound-healing and Transwell assays were used to detect migratory and invasive ability. EGFR-TKI sensitivity was evaluated by assessing the proliferation, clonogenic survival and metastatic capability of cancer cells with treatment with gefitinib. Methylated RNA immunoprecipitation (MeRIP) and RNA immunoprecipitation (RIP) analyses established the level of m6A modification present on the target gene and the protein's capability to interact with RNA, respectively. Single-sample gene set enrichment (ssGSEA) algorithm used to investigate levels of immune cell infiltration.
Results: Our study identified dual-specificity phosphatase 5 (DUSP5) as a novel and powerful predictor of adverse outcomes for LUAD by using public datasets. Functional enrichment analysis found that DUSP5 was positively enriched in EMT and transforming growth factor-beta (TGF-β) signaling pathway, a prevailing pathway involved in the induction of EMT. As expected, DUSP5 knockdown suppressed EMT via inhibiting the canonical TGF-β/Smad signaling pathway in in vitro experiments. Consistently, knockdown of DUSP5 was first found to inhibit migratory ability and invasiveness of LUAD cells in in vitro and prevent lung metastasis in in vivo. DUSP5 knockdown re-sensitized gefitinib-resistant LUAD cells to gefitinib, accompanying reversion of EMT progress. In LUAD tissue samples, we found 14 cytosine-phosphate-guanine (CpG) sites of DUSP5 that were negatively associated with DUSP5 gene expression. Importantly, 5'Azacytidine (AZA), an FDA-approved DNA methyltransferase inhibitor, restored DUSP5 expression. Moreover, RIP experiments confirmed that YTH N6-methyladenosine RNA binding protein 1 (YTHDF1), a m6A reader protein, could bind DUSP5 mRNA. YTHDF1 promoted DUSP5 expression and the malignant phenotype of LUAD cells. In addition, the DUSP5-derived genomic model revealed the two clusters with distinguishable immune features and tumor mutational burden (TMB).
Conclusions: Briefly, our study discovered DUSP5 which was regulated by epigenetic modification, might be a potential therapeutic target, especially in LUAD patients with acquired EGFR-TKI resistance.
Partial results of cooperation

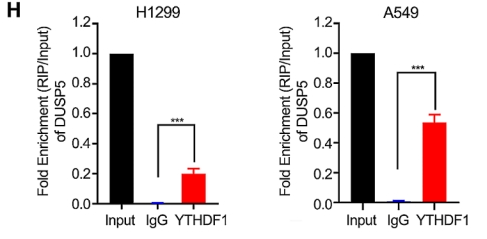
BersinbioTM cooperative technology:RIP
Original link:10.1186/s12935-024-03382-6